$title Parameterization of molecule T
$print 0
$equil ../QMdata/opt+freq.fchk
$forcefield gromacs example.top
$generate
$zero 1.d-12
$whess 5000. 2500.0
$geom
../QMdata/scan1.0.fcc 5.0 0.0 0.0 0.0 ; 1 2 3 4
../QMdata/scan1.30.fcc 5.0 0.0 0.0 0.0 -f
../QMdata/scan1.60.fcc 10.0 0.0 0.0 0.0 -f
../QMdata/scan1.90.fcc 5.0 0.0 0.0 0.0 -f
[...]
$end
$assign
1 = 2241.69 ; C1-C2
2 = 2143.14 ; C2-C2
34 = 732.30 ; C1-C2-C1
$end
$dependence 1.2
43 = 42*1.d0 ; C2-C2-C1-H1 = H1-C1-C2-C2
$end
$keepff 38 - 103
$LJassign
$scan joyce.scan1.dat
1 2 3 4 ; -180. 180. 1.
$end
$gracefreq joyce.molname_freqchk.agr
$gracetors molname_torschk
$title
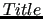 key simply sets a title for your job, that will appear in the joyce.molname.out output file.
This is an optional key. key simply sets a title for your job, that will appear in the joyce.molname.out output file.
This is an optional key.
$print
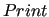 key sets the verbosity in the output log file.
It has to be followed from a number in the -1,4 range,
being -1 the less verbose mode, whereas 4 should be used in debugging sessions only.
This is an optional key (the default is 0). key sets the verbosity in the output log file.
It has to be followed from a number in the -1,4 range,
being -1 the less verbose mode, whereas 4 should be used in debugging sessions only.
This is an optional key (the default is 0).
$equil
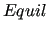 keyword is required.
It must be followed by the exact path pointing to the .fcc or
.fchk file, containing target T's optimized geometry and Hessian matrix. keyword is required.
It must be followed by the exact path pointing to the .fcc or
.fchk file, containing target T's optimized geometry and Hessian matrix.
In the above example, a GAUSSIAN16 .fchk file is located in QMdata directory and is named opt+freq.
$forcefield
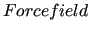 keyword is also required.
It specifies both format type and location (via its path) of the topology input file. As already noted, the supported input format is the gromacs [18] .top topology file software packages. keyword is also required.
It specifies both format type and location (via its path) of the topology input file. As already noted, the supported input format is the gromacs [18] .top topology file software packages.
In the above example the input topology file is located in the working directory and named example.top.
$generate
This keyword should be only employed in Step 0. When the $generate key is activated, the program does not perform any fitting and instead it creates a new text file, named generated.IC.txt, where all the internal coordinates of the target molecule are written in the GROMACS 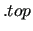 format and can be used as starting topology file in subsequent runs.
RIC's are suggested considering bond order and geometrical information retrieved from the QM optimized geometry.
Note that after release 3.0, the generated RICs for dihedrals are also automatically associated with the suggested model function, either harmonic or periodic, depending on the IC expected stiffness (see Section 7). format and can be used as starting topology file in subsequent runs.
RIC's are suggested considering bond order and geometrical information retrieved from the QM optimized geometry.
Note that after release 3.0, the generated RICs for dihedrals are also automatically associated with the suggested model function, either harmonic or periodic, depending on the IC expected stiffness (see Section 7).
$zero
This keyword is optional, and sets the lower limit of the numerical significance of the computed eigenvalues.
In other words, all eigenvalues lower than this threshold will be considered as null, and discarded.
It is optional, and its default is 1
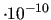 .
$whess .
$whess
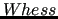 keyword is required for
performing any QMD-FF parameterization.
It sets the diagonal and off diagonal weights of the Hessian matrix elements to be used in the objective function I (see Section 7).
In the example above, such weights are set to standard values commonly used ion most JOYCE parameterizations.
Note that Hessian diagonal elements are normally weighted twice with respect to off-diagonal ones. keyword is required for
performing any QMD-FF parameterization.
It sets the diagonal and off diagonal weights of the Hessian matrix elements to be used in the objective function I (see Section 7).
In the example above, such weights are set to standard values commonly used ion most JOYCE parameterizations.
Note that Hessian diagonal elements are normally weighted twice with respect to off-diagonal ones.
$geom
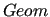 keyword is required for Step 2. This keyword is needed when
performing QMD-FF parameterizations that involve target molecules with flexible coordinates, for which QM relaxed torsional scans were carried out.
In these cases, this keyword reads the additional QM
files, corresponding to partial optimizations with the flexible IC constrained.
All these files have to be listed
in the $geoms field, which is ended by $end.
For each row, the path and filename of each .fchk file should be specified.
This is followed by four numbers, indicating the energy, gradient, diagonal and off diagonal Hessian weights to be applied to the objective function I.
Note that FIRA is applied to these RICS, and the quadruplet(s) of atoms defining
the involved torsion(s) is(are) specified afterwards (preceded by a ";").
Clearly, for each scan performed along a torsional coordinate, all sampled points (resulting in separate .fcc or .fchk files)
refer to the same quadruplet.
Starting from the second sampled point,
the quadruplet(s) complete declaration in the $geoms environment can be omitted
for the sake of brevity, and
substituted by the symbol "-f". This indicates that the FIRA declarations should be read from the previous line. keyword is required for Step 2. This keyword is needed when
performing QMD-FF parameterizations that involve target molecules with flexible coordinates, for which QM relaxed torsional scans were carried out.
In these cases, this keyword reads the additional QM
files, corresponding to partial optimizations with the flexible IC constrained.
All these files have to be listed
in the $geoms field, which is ended by $end.
For each row, the path and filename of each .fchk file should be specified.
This is followed by four numbers, indicating the energy, gradient, diagonal and off diagonal Hessian weights to be applied to the objective function I.
Note that FIRA is applied to these RICS, and the quadruplet(s) of atoms defining
the involved torsion(s) is(are) specified afterwards (preceded by a ";").
Clearly, for each scan performed along a torsional coordinate, all sampled points (resulting in separate .fcc or .fchk files)
refer to the same quadruplet.
Starting from the second sampled point,
the quadruplet(s) complete declaration in the $geoms environment can be omitted
for the sake of brevity, and
substituted by the symbol "-f". This indicates that the FIRA declarations should be read from the previous line.
In the example reported above, two torsional profiles are considered into the fitting.
The first dihedral is defined by atoms 1 to 4, and the energy, gradients and Hessian matrixes relative to all sampled points are retrieved from the GAUSSIAN16 scan1.*.fchk ( * = 0, 30, 60 and 90) files, located in the same directory as opt+freq.fchk file.
The second dihedral is defined by atoms 3-2-1-12, and the QM information
will be extracted from the files named scan2.*.fchk (* = 0, 30, 60 and 90) and again located in the G09 directory.
For all these conformations, only the energy will be considered in the fitting, as all other weights are set to 0. In particular, the weight will be 5 for all energies but those referring to files scan1.60.fchk and scan2.60.fchk, which are set to 10.
$assign
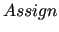 keyword is required for Step 2, as it allows for safely applying FIRA[1,21], by assigning all other harmonic QMD-FF constants to the values obtained in the previous Step 1, based on the Hessian matrix.
This keword can also be useful in Step 1, for instance when some of the force constant of the FF have to be constrained to a definite value, chosen by the user or taken from other FF's.
The list of such constants is declared in the $assign
field, where the IC number (consistent with the numbering found in the topology file and reported in the joyce. keyword is required for Step 2, as it allows for safely applying FIRA[1,21], by assigning all other harmonic QMD-FF constants to the values obtained in the previous Step 1, based on the Hessian matrix.
This keword can also be useful in Step 1, for instance when some of the force constant of the FF have to be constrained to a definite value, chosen by the user or taken from other FF's.
The list of such constants is declared in the $assign
field, where the IC number (consistent with the numbering found in the topology file and reported in the joyce. .out log file) is first declared, followed by a "=" symbol and the value of the force constant, relative to that IC, to be constrained.
Comments can be added after a ";" symbol, to explicitly declare the coordinate type one is dealing with. .out log file) is first declared, followed by a "=" symbol and the value of the force constant, relative to that IC, to be constrained.
Comments can be added after a ";" symbol, to explicitly declare the coordinate type one is dealing with.
Units are kJ/(mol Å ), kJ/(mol rad) and kJ/mol for stretching, bending and torsions, respectively. ), kJ/(mol rad) and kJ/mol for stretching, bending and torsions, respectively.
In the framed example of JOYCE3.0 input file reported above, three force constants are assigned.
As it can be deduced by the comments (indicated with the ";" symbol, the first two refers to stretching coordinates (IC number 1, between atoms labels C1 and C2
and IC number 4, between C2 and C2 types), while the last refer to a bending coordinate (IC number 34, defined by C1-C2-C3 sites). These force constants will not be fitted but are constrained to 2241.69 kJ/(mol Å), 2143.14 kJ/(mol Å), and 732.30 kJ/(mol rad), respectively.
$dependence
Some IC's may be equivalent, either for symmetry reasons or because a similar chemical behavior is expected.
In these cases it is desirable to describe such IC's with the same equilibrium distances and force constants.
In other words, given two equivalent IC's, the parameter values of the second are dependent on those of the first.
JOYCE can find dependencies automatically, based on symmetry
considerations.
A number between 0.5 and 1.0 should be specified as argument of the $dependence field, which sets JOYCEsensitivity to IC's equivalence: lower values correspond to less strict criteria, while when this number is set to 1.0 (default), only highly dependent coordinates are considered.
Values higher than 1.0 disable the automatic selection of dependent coordinates, and a list of all equivalent IC's may be given manually.
This can be done by specifying the number consistent with the numbering found in the topology file and reported in the joyce..out log file) of the dependent IC, followed by a "=" symbol, the number of the reference IC, a "*" symbol and the number (usually 1 for equivalent IC's) for which the force constant of the reference IC should be multiplied to give the one of the dependent IC.
In the framed example of JOYCE3.0 input file above-reported, the force constant of IC number 43 is forced during the fitting to take the same value of the one relative to IC 42.
The reason for this choice is noted in the comment: both dihedrals are defined by the same quadruplet of atom types.
Since 1.2 is indicated as sensitivity number, no other dependencies will be imposed.
$keepff
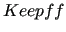 keyword is requisite for Step 2, i.e. when parameterizing flexible coordinates through model periodic functions.
As for the $assign field, also the equilibrium values ( keyword is requisite for Step 2, i.e. when parameterizing flexible coordinates through model periodic functions.
As for the $assign field, also the equilibrium values (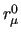 , ,
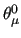 , etc, see Section 7) of the FF can be constrained to the value declared in the parameter input file.
The list of the RICs, for which the equilibrium values should be taken from the parameter file and not from the GAUSSIAN16 equilibrium geometry, is declared as argument of the $keepff
field.
Here, the IC number (consistent with the numbering found in the topology file and reported in the joyce..out log file) can be given separately (e.g. 3, 6, 12, 54) or grouped (e.g. 3 - 10).
While it is advisable to force JOYCE to read the , etc, see Section 7) of the FF can be constrained to the value declared in the parameter input file.
The list of the RICs, for which the equilibrium values should be taken from the parameter file and not from the GAUSSIAN16 equilibrium geometry, is declared as argument of the $keepff
field.
Here, the IC number (consistent with the numbering found in the topology file and reported in the joyce..out log file) can be given separately (e.g. 3, 6, 12, 54) or grouped (e.g. 3 - 10).
While it is advisable to force JOYCE to read the
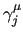 phase constants of the periodic functions, the equilibrium values of the "stiff" IC should be, as far as possible, taken from the QM equilibrium geometry. phase constants of the periodic functions, the equilibrium values of the "stiff" IC should be, as far as possible, taken from the QM equilibrium geometry.
In the example, the geometrical parameters related to IC number 38 to 103 will be taken from the example.top topology
file and not from the QM equilibrium geometry read from the
opt+frq.fchk GAUSSIAN16 file.
$LJassign
This keyword is optional, yet recommended in Step 2 when dealing with very flexible targets, which required the use of non-bonded intramolecular terms.
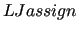 was introduced since release 3.0, and activates the so called "Route II" [28] to handle FIRA and internal LJ.
Concretely, with this keyword all intramolecular LJ interactions declared in the input topology file are included in the QMD-FF, and the torsional terms fitted while taking into account their contribution. was introduced since release 3.0, and activates the so called "Route II" [28] to handle FIRA and internal LJ.
Concretely, with this keyword all intramolecular LJ interactions declared in the input topology file are included in the QMD-FF, and the torsional terms fitted while taking into account their contribution.
$gracefreq
This keyword requires JOYCE3.0 to print a 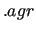 file, compatible with the popularXMGRACE graphic software,[27] which contains information on the quality of the Hessian fitting in terms of FF vs. QM vibrational frequencies and normal modes. file, compatible with the popularXMGRACE graphic software,[27] which contains information on the quality of the Hessian fitting in terms of FF vs. QM vibrational frequencies and normal modes.
$gracetors
Similar to the previous keyword and implemented since the 3.0 release, it allows to print an file containing information on the quality of the relaxed scan fitting.
JOYCE3.0 automatically produce separate files for each torsion considered in the following 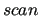 section. section.
$scan
When the $geom key is activated, i.e. when the target molecule has very flexible degrees of freedom, it may be desirable check the capability of the parameterized QMD-FF to reproduce the QM computed torsional profiles.
This can be done by comparing the QM energy curves, computed with respect of the minimum energy geometryfrom the files specified in
$geoms field (and collected in the qmscan.*.dat JOYCEoutput files), with those obtained with the parameterized FF.
The first argument of the $scan keyword is the name of the file in which the FF energy profile should be printed. This should be followed by the declaration of IC number for which an energy scan is requested and the specifications of the scan.
The IC's is identified by specifying its defining quadruplet (or triplet for bending) of atoms followed by
a ";" symbol.
The scan directive are given by
specifying the initial and final value that the IC should
assume during the scan and the required scan step.
The $scan keyword can be repeated for each
IC for which the scan is requested.
In the framed example file, only a scan is requested, to be printed in the working directory in a file named joyce.scan1.dat. The scan refers to the dihedral defined by atoms 1 to 4, and the FF energy will be computed for all values of this dihedral from -180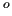 to 180 in steps of 1. to 180 in steps of 1.
Moreover, the next commands are not included in the above reported framed sample input file, however they activate optional useful features of the JOYCE program.
$rearr
It may happen that the atom numbering employed in the QM calculations differs from the one chosen in the parameter input file. In some cases it may be rather cumbersome to change the order atoms appear, since all the computed informations (Hessian matrices in .fchk and IC definitions in FF files) depend on atom numbering. The rearr command solves this problem by rearranging the atom numbering in the QM data.
$rearr 1 2 3 4 5 6 7 8 12 13 \
14 11 10 9
In the above example the numbering of the first eight atoms is unchanged, whereas atoms 12 to 14 are moved in positions 9 to 11, atom 11 is placed in 12
 positions, etc. positions, etc.
$UnitedAtom
This keyword introduces the UA approximation. The theory is described in some detail in section 7.2.4.
The UnitedAtom environment is closed by a
$end command. Between these keys all UA sites are specified by
indicating the number of the atom whose position will be the center of the UA site, followed by the numbers of all atoms that should be grouped to it.
An example is framed in the following.
$UnitedAtom
8 17 18
10 21 22
11 23 24 25
$end
Here atoms 17 and 18 are considered as a unique interaction site with atom 8, with the latter being the atom center of the new UA site. The same procedure is applied to atoms 21,22 and 10, whereas four atoms (23, 24, 25 and 11) are included in the third UA site.
$fitLJ
This key allows J OYCE to fit the Lennard-Jones force constants ( 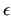 's) defined for non-bonded intramolecular interactions, as reported in equation ( 11).
It is important to note that for default LJ interactions are not fitted by JOYCE, but they are included as constants in the FF energy expressions.
Conversely, when the
fitLJ key is activated, all employed 's values may be parameterized, so the user needs to explicitly specify those values that he wants to keep constant in the assign section.
Note also that geometrical LJ values ( 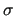 's) are always taken from the input FF parameter file.
$sep_el
This key allows JOYCE to be coupled with polarizable models or, more generally, with electrostatic models other than standard Coulomb interaction (3).
In this case, a second argument is needed in the
$equil section, indicating a path to a .fchk file containing the Hessian elements computed for only the electrostatic part of the Hamiltonian. This implies a slight modification of the merit functional (35). Further details can be found in JOYCE 2013 paper [29].
$mass
This key is followed by a number, which is used to set all atoms to a unique mass.
It is intended for particular applications and its standard use is deprecated.
$normal
This keyword sets the computational route to get vibrational normal modes.
With no arguments (default or without using the $normal keyword) JOYCE computes vibrational normal modes by working in a space orthogonal to the translational and rotational modes (by using the Graham-Schmidt orthogonalization).
Conversely, if zero is used as argument of the $normal keyword the normal modes are computed directly by analyzing the null eigenvectors.
$wfreq
This keyword should be followed by a scaling factor to empirically adjust the computed vibrational frequencies, to correct QM systematic errors when present.
$boltz
A Boltzmann weight can be introduced with this keyword, to weight the energies of the geometries considered.
Note that this is alternative to the weights assigned in the $geoms section, which is instead recommended.
|
|
|