The total energy (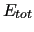 ) of a system of
) of a system of 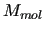 molecules in classical simulation is usually [5,6] computed as a sum of two contributions, namely
molecules in classical simulation is usually [5,6] computed as a sum of two contributions, namely
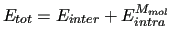 |
|
|
(1) |
where 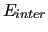 and
and
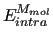 are the interaction energy among different molecules and the sum of the internal energy of each molecule.
In standard FFs, is computed as a sum of pairwise contributions among all the
are the interaction energy among different molecules and the sum of the internal energy of each molecule.
In standard FFs, is computed as a sum of pairwise contributions among all the 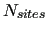 interaction sites used to model the system.
In particular,
interaction sites used to model the system.
In particular,
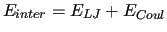 |
|
|
(2) |
where the long-range electrostatic term is
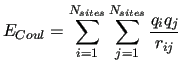 |
|
|
(3) |
whereas the short range 12-6 Lennard-Jones term is
![$\displaystyle E_{LJ} = \sum^{N_{sites}}_{i=1} \sum^{N_{sites}}_{j=1}
4 \epsilon...
...} {r_{ij} } \bigg )^{12} -
\bigg(\frac{ \sigma_{ij} } {r_{ij} } \bigg)^6 \bigg]$](img107.png) |
|
|
(4) |
where 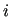 and
and 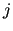 are interaction sites belonging to a pair of different molecules.
are interaction sites belonging to a pair of different molecules.
The total intramolecular term
is the sum of the molecular internal energies (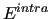 ) of all the
) of all the 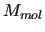 molecules composing the system,
i.e.
molecules composing the system,
i.e.
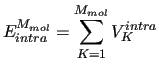 |
|
|
(5) |
where
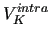 is the internal energy of molecule
is the internal energy of molecule 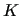 .
.
Given a model potential function
, the main goal of the JOYCE program is to find, with respect of reference QM computed data, the best parameters to represent the intramolecular energy for a chosen target molecule , hence parameterizing the intra-molecular term of a QMD-FF..