Starting .top file: n-Butane
; --------------
; 1) Inter-molecular part :
; --------------
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 3 no 1.0 0.0
[ atomtypes ]
; name mass charge ptype sigma(nm) epsilon (kJ/mol)
C1 12.0110 -0.18 A 0.350 0.27612
C2 12.0110 -0.12 A 0.350 0.27612
H1 1.0079 0.06 A 0.250 0.12555
H2 1.0079 0.06 A 0.250 0.12555
; ---------------
; 2) Intra-molecular part :
; ---------------
[ moleculetype ]
; Name nrexcl
but 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 C1 1 but C1 1 -0.180 12.0110
2 C2 1 but C2 2 -0.120 12.0110
3 C2 1 but C2 3 -0.120 12.0110
4 C1 1 but C1 4 -0.180 12.0110
5 H2 1 but H2 5 0.060 1.0079
[...]
14 H1 1 but H1 6 0.060 1.0079
[ bonds ]
; ai aj type r0 (nm) ks (kJ/(mol nm^2))
1 2 1 0.0 0.0 ; 1 C1-C2
2 3 1 0.0 0.0 ; 2 C2-C2
[ ... ]
1 14 1 0.0 0.0 ; 13 C1-H1
; ai aj ak type th0 (degr) kb (kJ/(mol rad^2)
1 2 3 1 0.0 0.0 ; 14 C1-C2-C2
2 3 4 1 0.0 0.0 ; 15 C1-C2-C2
[ ... ]
2 1 13 1 0.0 0.0 ; 36 C2-C1-H1
2 1 14 1 0.0 0.0 ; 37 C2-C1-H1
[ dihedrals ]
; ai aj ak al type gam kd (kJ/mol) n
1 2 3 4 1 0.0000 0.0 0 ; 38 C1-C2-C2-C1
1 2 3 4 1 180.0000 0.0 1 ; 39 C1-C2-C2-C1
1 2 3 4 1 90.0000 0.0 2 ; 40 C1-C2-C2-C1
1 2 3 4 1 0.0000 0.0 3 ; 41 C1-C2-C2-C1
1 2 3 4 1 0.0000 0.0 4 ; 42 C1-C2-C2-C1
12 1 2 3 1 0.0000 0.0 3 ; 43 H1-C1-C2-C2
2 3 4 9 1 0.0000 0.0 3 ; 44 C2-C2-C1-H1
[ pairs ]
; ai aj f_qq qi qj sigma (nm) epsilon (kJ/mol)
; 1-4 C1-C1
1 4 2 0.0 0.0 0.0 0.3700 0.1
; ai aj
1 2
1 3
[...]
13 14
; --------------
; 3) System definition :
; --------------
; Name
n-butane
[ molecules ]
; Compound #mols
but 1
Intermolecular section
The intermolecular section has always to be the first in a .top file.
It contains the atom labels definition and it is made up of two environments.
[defaults]
The [defaults] environment defines the potential functions used to compute non-bonded interactions, reported in equation (11) in Section 7.
The outline of this section has always to be the following:
Variable $1 defines the form of the non-bonded model potential function, and $1 = 1 sets the LJ form, shown in equation (4).
Variable $2 sets the combination rules to be used to compute
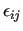 and and
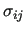 from the single site values from the single site values
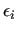 and and
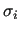 : $2 = 2 selects Lorentz-Berthelot mixing rules, while OPLS ones are chosen by setting $2 = 3.
The third variable ($3 = yes/no ) forces GROMACS to compute non-bonded intramolecular interactions by using the same
and
given for the intermolecular ones.
Variables $4 and $5 define the scaling factors that will be respectively applied to LJ and Coulomb terms of the non-bonded intramolecular interactions. : $2 = 2 selects Lorentz-Berthelot mixing rules, while OPLS ones are chosen by setting $2 = 3.
The third variable ($3 = yes/no ) forces GROMACS to compute non-bonded intramolecular interactions by using the same
and
given for the intermolecular ones.
Variables $4 and $5 define the scaling factors that will be respectively applied to LJ and Coulomb terms of the non-bonded intramolecular interactions.
Note that JOYCE ignores the latter three variables (but they are copied and pasted into the final output topology file), so variable $3 should be switched off all the time, to avoid confusion.
If intramolecular non-bonded LJ interaction are to be used within the JOYCE3.0 procedure, the intramolecular
and
values, together with the chosen scaling factors, should be separately defined in the [pairs] section (see the following).
Conversely, if the $generate key is activated, the LJ intramolecular parameters will be automatically created through the appropriate mixing rules form the
and
values read from the [atomtypes] section, illustrated in the next section.
In the sample file above,
OPLS combination rules are chosen for the n-butane molecule's inter-molecular term, whereas LJ intramolecular pairs are specifically defined in the [atomtypes] section (vide infra).
[atomtypes]
In this environment, all atom types of the adopted model are defined, by specifying for each type 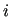 its mass, charge and
and
intermoilecular LJ parameters.
It is important to stress that the JOYCE procedure allows for defining any number of atom types, ranging from the few provided by general purpose FFs to more specific descriptions (dictated by symmetry or chemical equivalence).
The user is encouraged to visit JOYCE3.0 website[13] for a gallery of different examples.
The outline of the [atomypes] section is the following:
Variable $1 defines the atom type label, $2 its mass in
g its mass, charge and
and
intermoilecular LJ parameters.
It is important to stress that the JOYCE procedure allows for defining any number of atom types, ranging from the few provided by general purpose FFs to more specific descriptions (dictated by symmetry or chemical equivalence).
The user is encouraged to visit JOYCE3.0 website[13] for a gallery of different examples.
The outline of the [atomypes] section is the following:
Variable $1 defines the atom type label, $2 its mass in
g mol mol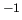 , $3 its charge in , $3 its charge in
 e. Variable $4 defines the kind of particle atom type refer to: in GROMACS three ptypes are possible, namely atoms ($4 = A), shells ($4 = S) or virtual sites ($4 = V).
The last two variables, $5 and $6, set the values of the atom type LJ parameters
and
, in nm and kJ/mol, respectively.
In the framed sample file, three atom types are defined namely H, C1 and C2 for Hydrogen, CH e. Variable $4 defines the kind of particle atom type refer to: in GROMACS three ptypes are possible, namely atoms ($4 = A), shells ($4 = S) or virtual sites ($4 = V).
The last two variables, $5 and $6, set the values of the atom type LJ parameters
and
, in nm and kJ/mol, respectively.
In the framed sample file, three atom types are defined namely H, C1 and C2 for Hydrogen, CH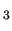 and CH and CH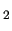 carbon atoms, respectively. carbon atoms, respectively.
Intramolecular section
The molecular topology is defined here in a number of subsections, which are discussed in the following.
[moleculetype]
This subsection is required, because, like for atom types, any molecule has to be specified in GROMACS by a label.
In the intramolecular section, target T's molecule-type is defined in the [moleculetype] subsection.
Variable $1 is used to identify molecular type by a string which can be up to 10 characters long, while $2 defines the number of bonds which have to separate two sites for the intramolecular non-bonded interactions to be computed.
In the example file framed at the beginning of this Section but is the label assigned to the n-butane molecule, whereas $2 = 3 stands for excluding non-bonded interactions between atoms that are closer than 3 bonds.
[atoms]
The [atoms] subsection is the most important environment, as it defines the molecule, matching each atom with its atom-types, defined in [atomtypes].
In JOYCE parameterizations this section must follow the [moleculetype] subsection, because all other subsections use the order of sites declared here.
[atoms]
$1$2$3$4$5$6$7$8
|
Variable $1 specifies the atom number, while $2 specifies its atom type.
Arguments $3 and $4 are used by GROMACS to specify the residue or molecule to which the atoms belong, by giving its number and label, respectively.
Variable $5 specifies the atom name, while $6 indicates the charge group (see GROMACS manual for further details).
Finally, last two variables set atomic charge and mass, which can be varied by the user with respect to the one given in the [atomtypes] environment.
In the JOYCE program, it is fundamental that the order of the atoms (specified by $1) matches exactly the order used in the QM calculations (this in all .fcc or .fchk files).
Although severe tests are automatically performed by JOYCE3.0 to verify this feature, the user should always control the numbering of atoms in both files.
In the example reported in frame, the label C1 is assigned to Carbon atoms of the CH groups, while C2 label indicate internal Carbons.
Atoms 4 to 14 are all Hydrogen atoms, corresponding to a unique atom type, H.
[bonds]
Within the [bonds] environment GROMACS allows for several distinct types of bonded pair interactions, whose detailed description can be found in the online manual. [18]
At the moment only harmonic stretching is implemented in JOYCE3.0, so only these interactions will be described.
Indeed, each harmonic bond between two sites is defined by writing the following line.
[bonds]
$1$2$3$4$5
|
Variables $1-$5 are defined according to equation (7) in Section 7, where $1 and $2 define the atom numbers (referred to the ordering given in the [atoms] environment) of the pair 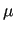 involved in the bond, $4 is the equilibrium distance involved in the bond, $4 is the equilibrium distance 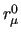 in nm and $5 the stretching force constant in nm and $5 the stretching force constant 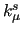 in kJ/(mol nm in kJ/(mol nm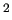 ).
Finally argument $3 indicates the type of function to be used for the stretching potential: $3 = 1 sets the harmonic form.
In the sample .top file, framed at the beginning of this section, the definitions of selected bonds for the butane molecule are shown.
As noted in the comment, the first bond refers to bond between the first C1 and C2 atoms, while the second line describes the C2-C2 IC.
The last line describes a C1-H type bond, between atoms 1 and 14.
The IC numbering in the JOYCE procedure is thus taken from this ordering:
distance between atoms 1 and 2 (C1-C2) will be IC number 1, distance between atoms 2 and 3 (C2-C2) IC number 2, distance between 3 and 4 (C2-C1) number 4, and so on. Note that in the .top file produced by JOYCE3.0 the IC numbering is explicitly declared in the comment as reported the template above. ).
Finally argument $3 indicates the type of function to be used for the stretching potential: $3 = 1 sets the harmonic form.
In the sample .top file, framed at the beginning of this section, the definitions of selected bonds for the butane molecule are shown.
As noted in the comment, the first bond refers to bond between the first C1 and C2 atoms, while the second line describes the C2-C2 IC.
The last line describes a C1-H type bond, between atoms 1 and 14.
The IC numbering in the JOYCE procedure is thus taken from this ordering:
distance between atoms 1 and 2 (C1-C2) will be IC number 1, distance between atoms 2 and 3 (C2-C2) IC number 2, distance between 3 and 4 (C2-C1) number 4, and so on. Note that in the .top file produced by JOYCE3.0 the IC numbering is explicitly declared in the comment as reported the template above.
[angles]
As for stretching, more than one functional form is implemented in GROMACS to describe angle bending. [18]
However the following discussion will be limited to harmonic form.
A harmonic angle bending interaction is defined in the [angles] section according to
[angles]
$1$2$3$4$5$6
|
where the parameters $1-$6 indicate the triplet of atoms defining the angle ($1-$3), the equilibrium angle
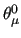 in degrees ($5)
and the bending force constant in degrees ($5)
and the bending force constant 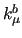 in kJ/(mol rad) ($6) that enter in equation (8) in Section 7.
As for stretching, argument $4 indicates the type of function to be used for the bending potential: $4 = 1 sets the harmonic form.
In the sample file some of the butane angles are defined. In such way, bending IC are added to the stretching IC included in the FF by defining them in the previous bond environment.
IC numbering just increases sequentially: if for instance 13 is the number of the last defined bond, the first angle here reported will be IC number 14, and so on. in kJ/(mol rad) ($6) that enter in equation (8) in Section 7.
As for stretching, argument $4 indicates the type of function to be used for the bending potential: $4 = 1 sets the harmonic form.
In the sample file some of the butane angles are defined. In such way, bending IC are added to the stretching IC included in the FF by defining them in the previous bond environment.
IC numbering just increases sequentially: if for instance 13 is the number of the last defined bond, the first angle here reported will be IC number 14, and so on.
[dihedrals]
GROMACS provides several functional forms for the description of the potential with respect to a dihedral angle. [18]
In particular, both harmonic and Fourier forms, defined respectively in equation (9) and
(10) in Section 7, are supported and have to be defined in the same[dihedrals] section according to the following format:
 - Stiff dihedrals: each harmonic dihedral is given by a line in the following form:
[dihedrals]
$1$2$3$4
$5$6$7
|
Variable $5 is set to 2 to activate harmonic torsions.
All other variable ($1-$4 and $6-$7) refer to equation (9) and are are defined in the usual way: parameters $1-$4 indicate the atom quadruple which defines the dihedral
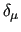 , $6 the equilibrium dihedral angle , $6 the equilibrium dihedral angle
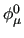 in degrees and $7 the harmonic torsion force constant in degrees and $7 the harmonic torsion force constant 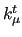 in kJ/(mol rad). in kJ/(mol rad).
- Soft dihedrals: the parameters defining the Fourier-like torsional potentials are defined as:
[dihedrals]
$1$2$3$4
$5$6$7$8
|
At difference with harmonic torsions, variable $5 is set to 1 to activate Fourier like torsions.
An anharmonic dihedral potential has to be defined in terms of a Fourier series with an, in principle, arbitrary number of cosine-terms.
The number of terms specifying the interaction for the site quadruple (
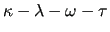 ), where ( ), where (
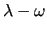 ) represents the central bond, is therefore also technically not restricted.
With reference to equation (10), arguments $1-$4 indicate, as in the previous case, the atom quadruplet which defines the dihedral.
Variables $6-$8 set the values for the phase angle ) represents the central bond, is therefore also technically not restricted.
With reference to equation (10), arguments $1-$4 indicate, as in the previous case, the atom quadruplet which defines the dihedral.
Variables $6-$8 set the values for the phase angle
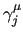 in degrees ($6),
the force constant (in kJ/mol) in degrees ($6),
the force constant (in kJ/mol)
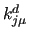 ($7) and the multiplicity ($7) and the multiplicity 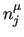 (($8) for the (($8) for the 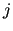 -th term in the Fourier series. -th term in the Fourier series.
Please note that if both harmonic and anharmonic forms have to be considered
(usually referred to different dihedrals) only one [dihedrals] environment is necessary, since both function types are defined by variable $5.
In the butane .top example file, all defined dihedrals are anharmonic.
Note that 5 cosine terms are used to describe the potential function for the dihedral formed by the four carbon atoms (1-4), while only one function is employed for the methyl dihedrals.
Please note that, despite 5 cosine terms are used for the potential function (see equation (10), only one IC arises from the same atom quadruplet.
In other words, only three new IC (dihedrals) are added to the IC list in the aforementioned example.
[pairs]
In almost all general purpose force-fields, [8,14,7,10,30] the
intramolecular non-bonded interactions are automatically included in the FF by accounting for the interactions among all atoms of the target molecule, except those that are less than three bonds away.
More specifically, the interactions within all considered pairs are computed through the usual sum of LJ and Coulomb terms, employing as such the same parameters defined for intermolecular interactions. The only exception being the 1-4 terms, that are usually scaled by some empirical factor.
However, GROMACS enables any intramolecular interaction to be modified specifically.
This feature is exploited by JOYCE in order to:
- select only specific atom pairs to interact, while excluding all the rest
- employ for such pairs user-defined LJ parameters
- turn off all charge-charge interaction within the molecule if not necessary (as in most neutral molecules)
All such specifics are given in the [pairs] section as follows:
[pairs]
$1$2$3$4$5
$6$7$8
|
where $1 and $2 are the numbers of the involved pair, variable $3 sets the functional form to be employed:
$3 = 1 uses only LJ term of equation (11), while $3 = 2 also adds Coulomb contributions.
In the former case ($3 = 1), $4 sets the value of
in nm and $5 the force constant
in kJ/mol.
Conversely, when $3 = 2, $4 indicates the linear scaling factor to apply to charge-charge interactions, $5 and $6 are the charges assigned to $1 and $2 atoms.
Finally $4 sets the value of
in nm and $5 the force constant
in kJ/mol.
It is important to underline once more that LJ parameters (and charges) concerning intramolecular interactions are defined here and may in principle differ from the ones employed for intermolecular interaction and defined in the [atoms] section.
Note also that in the standard JOYCE3.0 procedure, the charges (for intra-molecular interactions) are set to zero and hence not employed.
Nonetheless, the default topology created by JOYCE through the $generate command (see Section 5) lists all possible intramolecular pairs, yet assigning their parameters by applying Lorentz-Berthelot combination rules on the intermolecular parameters defined for each atom.
In the butane framed sample file reported at the beginning of this section, only a single non-bonded intramolecular IC is activated. The selected pair considers only the two methyl carbons, which interact through the sole LJ terms, with a user defined 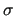 of 3.7Å and of 3.7Å and 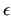 of 0.1 KJ/mol. of 0.1 KJ/mol.
[exclusions]
To consistently apply JOYCE3.0 default parameterization route [28] it is necessary to first switch off all intramolecular non-bonded interactions explicitly, and then declare only the selected active pairs through the [pairs] section as detailed above.
For this purpose all pairs have to be declared in the [exclusions] sections as follows:
[exclusions]
$1$2
$1$3
$1$4
$(N-1)$N
|
It is important to notice that the GROMACS topology format is very flexible in this regard, allowing for specific selection of interacting pairs through the combined use of the ![$ [pairs]$](img65.png) and and
![$ [exclusions]$](img66.png) environments.
It should be noticed that the environments.
It should be noticed that the 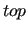 format follows strict priority rules in which all interactions defined in the section are overwritten to the
list.
Moreover, the user should avoid the automatic generation of pairs (in the format follows strict priority rules in which all interactions defined in the section are overwritten to the
list.
Moreover, the user should avoid the automatic generation of pairs (in the
![$ [ defaults]$](img68.png) section), as the interactions defined therein are created by the application of mixing rules to the inter-molecular parameters, and errouneously summed up to the ones defined in the section. section), as the interactions defined therein are created by the application of mixing rules to the inter-molecular parameters, and errouneously summed up to the ones defined in the section.
Couplings
In order to increase the flexibility of the FF, we have also adopted additional functional forms that accounts for couplings among different kinds of internal coordinates. Such couplings have revealed well adapted to describe the fluctuations in conjugation effects as the conjugated chains deviate from planarity[21].
The functional forms of the couplings terms are described in Section 7.1.3 (see Eqs. 20, 21 and 22), and they have been implemented both in the fitting procedure of JOYCE and in GROMACS MD engine. Namely, we maintain our own branch of GROMACS , where these potential forms are available[31],which accepts the following additional entries in the topology file.
[bo_di]
This entry defines parameters and the atoms involved in the coupling terms between bonds and flexible dihedrals (Eq. 20), in the following form:
[bo_di]
$1$2$3$4
$5$6$7
$8$9$10
|
Variables $1 and $2 define the bond () and $3 to $6 the dihedral angle (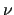 ). $7 is the function type (only one possible choice, ). $7 is the function type (only one possible choice, 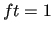 ) and remaining elements define the parameters of the function: $8 is the reference bond length (), $9 is the phase of the dihedral part ( ) and remaining elements define the parameters of the function: $8 is the reference bond length (), $9 is the phase of the dihedral part (
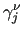 ), $9 is the force constant ( ), $9 is the force constant (
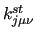 ) and $10 the multiplicity ( ) and $10 the multiplicity (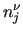 ). Note that different multiple entries may apply for a given couple of bond and dihedral, each with different multiplicity, which allows to define a Fourier-like series to determine the shape of the coupling. ). Note that different multiple entries may apply for a given couple of bond and dihedral, each with different multiplicity, which allows to define a Fourier-like series to determine the shape of the coupling.
[an_di]
This entry defines parameters and the atoms involved in the coupling terms between angles and flexible dihedrals (Eq. 21), in the following form:
[an_di]
$1$2$3$4
$5$6$7
$8$9$10$11
|
The variables follow similarly to [bo_di] entry: $1 to $3 define the angle () and $4 to $7 the dihedral angle (). $8 is the function type (only one possible choice, ) and remaining elements define the parameters of the function: $9 is the reference angle (
), $10 is the phase of the dihedral part (
), $10 is the force constant (
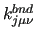 ) and $11 the multiplicity (). Again, it is possible to define a Fourier-like series to determine the shape of the coupling through multiple entries with different multiplicity. ) and $11 the multiplicity (). Again, it is possible to define a Fourier-like series to determine the shape of the coupling through multiple entries with different multiplicity.
[di_di]
This entry defines parameters and the atoms involved in the coupling terms between rigid dihedrals and flexible dihedrals (Eq. 22), in the following form:
[di_di]
$1$2$3$4
$5$6$7
$8$9$10
$11$12
|
In keeping with the two sections described above, each entry contains the variables: $1 to $4 define the rigid dihedral () and $5 to $8 the dihedral angle (). $9 is the function type (only one possible choice, ) and remaining elements define the parameters of the function: $10 is the reference value of the rigid (harmonic) dihedral (
), $11 is the phase of the dihedral part (
), $11 is the force constant (
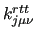 ) and $12 the multiplicity () with the possibility to define a Fourier-like series to determine the shape of the coupling through multiple entries with different multiplicity. ) and $12 the multiplicity () with the possibility to define a Fourier-like series to determine the shape of the coupling through multiple entries with different multiplicity.
System section
This section is only used by GROMACS during MD and ignored by JOYCE (but copied and pasted into the final output .top file).
It contains a title for the simulated system and the number of molecules for each species in the simulation.
In the framed sample file, the system is simply named 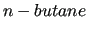 and only one molecule is expected. and only one molecule is expected.
It is important to stress that this section allows to extend the specific QMD-FF refined by JOYCE from gas-phase simulations to condensed phase systems.
For instance, bulk liquids mad up of 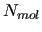 target molecules or solute-solvent systems, where solute target T is surrounded bu one or more different species.
In the case of systems with the same kind of molecules, only their number should be specified in the molecules section. This allows GROMACS to use the QMD-FF description for each of them.
Conversely, if target T has to be solvated, a new intramolecular section should be prepared for the solvent and appended to the one created by JOYCE. target molecules or solute-solvent systems, where solute target T is surrounded bu one or more different species.
In the case of systems with the same kind of molecules, only their number should be specified in the molecules section. This allows GROMACS to use the QMD-FF description for each of them.
Conversely, if target T has to be solvated, a new intramolecular section should be prepared for the solvent and appended to the one created by JOYCE.
|
|
|