 Figure:
Figure:
|
|
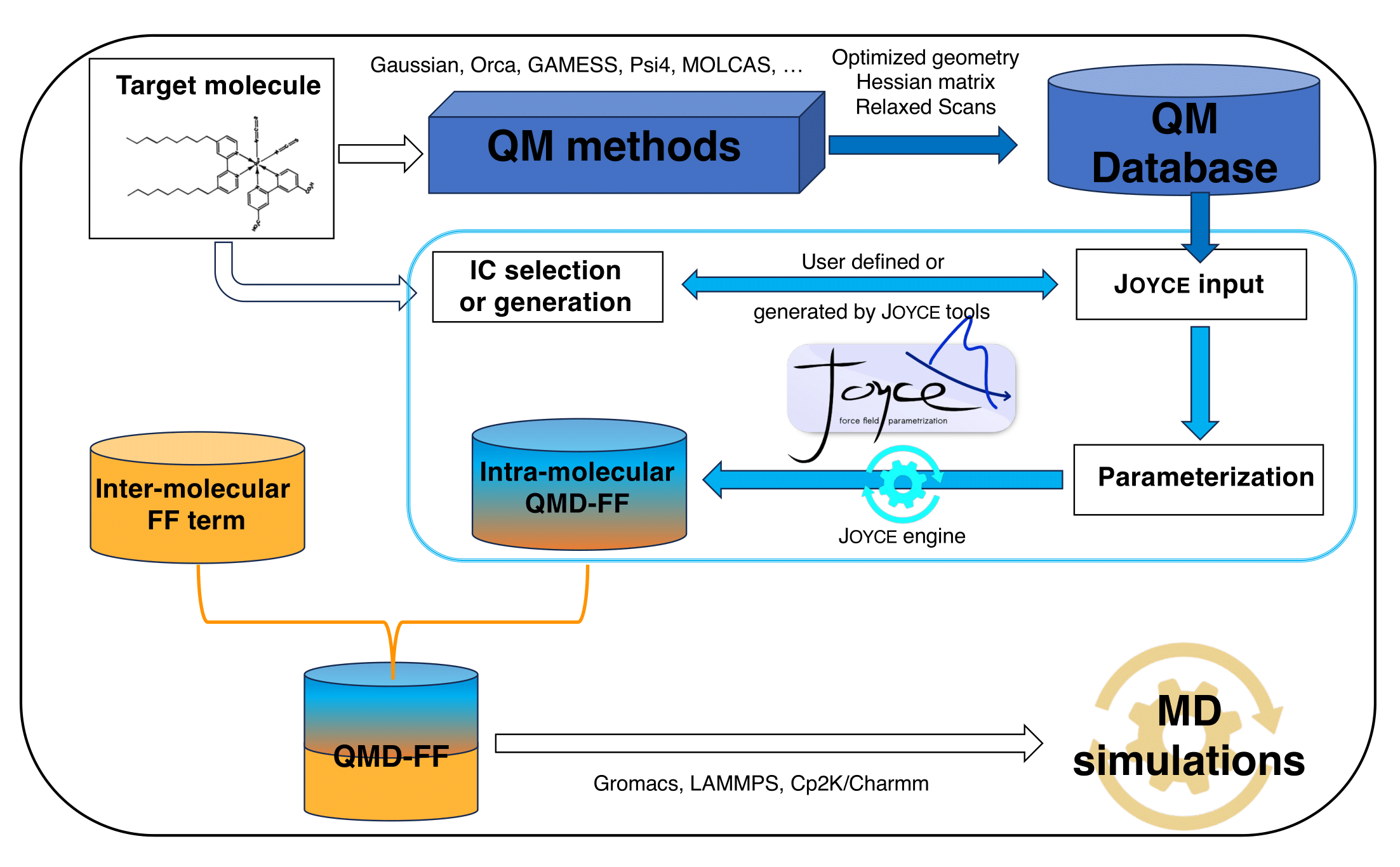
Starting from
T's chemical structure,
the JOYCE code is devised to simultaneously derive all parameters of a specific intramolecular FF term (see Section
7) from QM data purposely calculated on the target molecule.
Three kinds of information are required by J
OYCE to carry out the parameterization, each stored in a separate file.
The primary input file contains all the main instructions and specifics to run J
OYCE, and is discussed in some detail in Section
5.1.
Prior to parameterization (see top panels of Figure
1), the structure and flexibility of
T are first investigated at a proper QM level. This can be based on density functional theory (DFT), its time dependent extension (TD-DFT) or a wave-function (WF) based methods as HF, MP2 or CASPT2.
The computed QM descriptors are then stored in a database (see Section
3.3), which serves as J
OYCE's secondary input.
Briefly, J
OYCE3.0 can then be used in two ways:
 )
)- run a QMD-FF parameterization based on the collected QM database in terms of a well defined set of RICs and associated model potential functions, which is given in the third input file containing such molecular topology as detailed in Section 3.4;
 )
)- generate a proper collection of RICs and connected model functions (based on the QM connectivity), and store all information in a output topology file, to be used in a successive JOYCE parameterization run.
Once the definition of the IC set has been read and the QM data retrieved, J
OYCE3.0 is able to perform the parameterization, according to the theory reported in Section
7.
The final output of a J
OYCE parameterization consists in a complete collection of ICs, associated model functions and related FF parameters, stored in the output topology file as discussed in Section
3.5.
The latter can be used as such for gas phase simulations or completed with supplementary information concerning the inter-molecular parameters (which can be for instance taken from literature databases or refined on the basis of additional QM descriptors[
22,
3,
23]). This step makes QMD-FF suitable for condensed phase simulation containing molecule
T.