$assign
1 = 2254.5152345 C1-C2
2 = 2130.5117241 C2-C2
3 = 2254.5152345 C1-C2
4 = 3100.0797022 C1-H1
[...] 37 = 313.3362781 H1-C1-H1
38 = -1.9686612 C1-C2-C2-C1_n=0
39 = 4.3993865 C1-C2-C2-C1_n=1
40 = 1.7642241 C1-C2-C2-C1_n=2
41 = 7.5582350 C1-C2-C2-C1_n=3
42 = 0.1556409 C1-C2-C2-C1_n=4
43 = 2.1534692 H1-C1-C2-C2_n=3
44 = 2.1534692 H1-C1-C2-C2_n=3
45 = 2.1534692 H1-C1-C2-C2_n=3
46 = 2.1534692 H1-C1-C2-C2_n=3
47 = 2.1534692 H1-C1-C2-C2_n=3
48 = 2.1534692 H1-C1-C2-C2_n=3
$end
Although this procedure may seem a bit cumbersome, it can be very useful in the case of a well-different set of coordinates, such as stiff and soft IC. In this case it may be useful to fit in a first run all IC, only on the base of the optimized geometry and Hessian. Since the information used is insufficient to accurately characterized soft coordinates, a second run is necessary. In this second run, QM scans on soft IC are also read by JOYCE. Thus, to simplify the fitting procedure, the second run (i.e. the one with the QM scan data) can be performed keeping all stiff constants constrained to the values optimized in the first run. This is done by pasting into the second input file the assign.dat produce by JOYCE in the first run, after removing all soft IC from the list therein. Further details can be found by copnsulting the tutorials availabel at the JOYCE website.[13] It is worth noticing that, despite JOYCE adds at the end of the assigned list also the non-bonded intramolecular constants (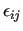 ,
,
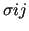 and the product
and the product
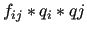 ), the LJ parameters and the atomic charges are not optimized by the
default settings of the program. These should be considered as assigned parameters, even if they do not appear in the assigned list.
The
parameters can be varied in the fitting procedure if the fitLJ
key is activated in the JOYCEinput. In this case, those
that the user wants to keep constrained, should be explicitly listed in the assign
environment.
), the LJ parameters and the atomic charges are not optimized by the
default settings of the program. These should be considered as assigned parameters, even if they do not appear in the assigned list.
The
parameters can be varied in the fitting procedure if the fitLJ
key is activated in the JOYCEinput. In this case, those
that the user wants to keep constrained, should be explicitly listed in the assign
environment.
1 = 2254.5152345 C1-C2
2 = 2130.5117241 C2-C2
3 = 2254.5152345 C1-C2
4 = 3100.0797022 C1-H1
[...] 37 = 313.3362781 H1-C1-H1
38 = -1.9686612 C1-C2-C2-C1_n=0
39 = 4.3993865 C1-C2-C2-C1_n=1
40 = 1.7642241 C1-C2-C2-C1_n=2
41 = 7.5582350 C1-C2-C2-C1_n=3
42 = 0.1556409 C1-C2-C2-C1_n=4
43 = 2.1534692 H1-C1-C2-C2_n=3
44 = 2.1534692 H1-C1-C2-C2_n=3
45 = 2.1534692 H1-C1-C2-C2_n=3
46 = 2.1534692 H1-C1-C2-C2_n=3
47 = 2.1534692 H1-C1-C2-C2_n=3
48 = 2.1534692 H1-C1-C2-C2_n=3
$end
Although this procedure may seem a bit cumbersome, it can be very useful in the case of a well-different set of coordinates, such as stiff and soft IC. In this case it may be useful to fit in a first run all IC, only on the base of the optimized geometry and Hessian. Since the information used is insufficient to accurately characterized soft coordinates, a second run is necessary. In this second run, QM scans on soft IC are also read by JOYCE. Thus, to simplify the fitting procedure, the second run (i.e. the one with the QM scan data) can be performed keeping all stiff constants constrained to the values optimized in the first run. This is done by pasting into the second input file the assign.dat produce by JOYCE in the first run, after removing all soft IC from the list therein. Further details can be found by copnsulting the tutorials availabel at the JOYCE website.[13] It is worth noticing that, despite JOYCE adds at the end of the assigned list also the non-bonded intramolecular constants (